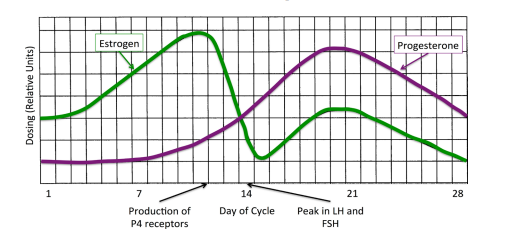
Abstract In recent years, Alzheimer’s disease has been clearly linked to the degradation of microtubules and microtubule-associated tau (τ) and β-amyloid (βA) proteins. Through an examination and evaluation of current literature, we assess the possible effects of the steroid hormones on τ hyperphosphorylation and the regulation of βA proteins and their influence on Alzheimer’s dementia and memory loss. We present a mechanism by which Alzheimer’s cases may be reduced or perhaps even prevented through the use of non-synthetic, steroid hormones prescribed in a cyclic dosing schedule that mimics the rhythmic, escalating and descending production normally observed in a reproductive female body. Given the ability of estrogen to prevent τ hyperphosphorylation and increase metabolism of the βA precursor protein, we propose the possibility of controlling both protein cycles through the exogenous application of estrogen and progesterone may help those patients with active disease as well as prevent the onset of Alzheimer’s and other neurodegenerative diseases. Keywords Hormones, Estradiol, Progesterone, Bio-Memtic, Treatment 1. Introduction Dementia is a chronic and persistent mental disorder that is typically marked by diminished memory, personality changes, and impaired reasoning [1]. A common form of the disease associated with dementia is Alzheimer’s disease (AD), which is characterized by neuronal cell loss, amyloid plagues, and vascular damage caused by plaque deposition [2] [3] [4] [5]. While all these factors can describe AD, the underlying cause remains an unknown. Since the first diagnosis of AD in 1907, there have been a number of theories How to cite this paper: Wiley, T.S. and Haraldsen, J.T. (2017) Cyclic E2 and P4 on Alzheimer’s Disease Pathways. Advances in Alzheimer’s Disease, 6, 32-44. https://doi.org/10.4236/aad.2017.61003 Received: February 26, 2017 Accepted: March 28, 2017 Published: March 31, 2017 Copyright © 2017 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/ Open Access T. S. Wiley, J. T. Haraldsen 33 about the cause and propagation of the disease [6] [7] [8] [9]. However, the underlying factor or cause has never been discovered. Alzheimer’s disease is a chronic neurodegenerative disease that affects an ever-increasing number of the elderly population [6]. With current healthcare costs increasing dramatically, there are a significant number of research groups and organizations examining mechanisms and causes in an attempt to understand and reduce the number of cases. In 2012, R. A. Armstrong presented an extensive review of the many theories about Alzheimer’s [6], where the possible causes were broken into eight main categories: aging, degeneration of anatomical pathways, environmental factors, genetics, mitochondrial dysfunction, vascular factors, immune system dysfunction, and infectious agents. The main issue for these theories is that the clinical data makes it difficult to determine whether they are primary causes or secondary effects from another more elusive problem [6] [7] [8] [9]. However, they do provide distinct pathologies that help to pinpoint the potential principal cause. The large number and wide range of possible candidates demonstrate the multifaceted range of factors and components that clinically affect Alzheimer’s patients. Therefore, in this manuscript, we look towards connecting all of these aspects to one overarching possibility. Recent evidence has demonstrated that the microtubule-associated protein tau (τ) and β-amyloid (βA) protein are linked to Alzheimer’s as well as other neurodegenerative diseases [10] [11] [12]. The τ protein stabilizes microtubules and is present in neurons located in the central nervous system [13] [14]. Alzheimer’s disease is characterized by the breakdown and loss of stability in the microtubules, which has indicated that the τ proteins have malfunctioned in some way to make them ineffective [11]. The impairment of the τ protein can be attributed to the hyperphosphorylation of the protein [12], which results in the tangling of various filaments that affect many neuromechanical pathways and can lead to multiple neurological disease states, including AD. The most widely studied protein is β-amyloid (βA), due to its connection to the formation of plaques observed in AD patients. However, the function of βA proteins remains fairly unknown. Although, it is suspected that they work to regulate neural plasticity and synapse formation [15]. However, the amino acid residues produced by βA helps to create βA plaques (βAP). The combination of neurofibril tangles due to τ hyperphosphorylation and βAP are both known to contribute to Alzheimer’s disease [12]. This occurs when βAP residues produce inflammation and transient ischemic attacks (TIAs) around affected areas of neural tissue. Since two-thirds of Americans with Alzheimer’s disease are women, the steroid hormone contributions to protein production of τ through gene expression controlled by the hormonal fluctuations of the menstrual cycle may be related to steroid hormones [16] [17]. Therefore, through an evaluation of current literature, we assess the possible effects of the steroid hormones (specifically estrogen and progesterone) on τ hyperphosphorylation and βA production and their influence on Alzheimer’s, dementia, and memory loss. We present a hypothesis that Alzheimer’s cases may be significantly reduced or even prevented with the T. S. Wiley, J. T. Haraldsen 34 use of non-synthetic, bio-identical steroid hormones prescribed in a cyclic dosing schedule that mimics the rhythmic, escalating and descending production created in a young reproductive female body. Given the ability of estrogen to prevent τ hyperphosphorylation and regulate βA precursor protein, as well as the overall effect of all steroid hormones on gene expression and the inflammatory process, the capability of controlling the expression of τ and βA through the exogenous application of estrogen and progesterone has the potential to help those patients with active disease symptoms as well as prevent the onset of Alzheimer’s. The general thesis of this article is to demonstrate that the loss of steroid hormones at menopause, specifically estrogen and progesterone, may provide a critical link between hyperphosphorylation of the τ proteins and the onset of Alzheimer’s disease in women. A similar mechanism may also exist in for men when the conversation of testosterone to estrogen falls off later in life. However, the various interactions and biomechanisms remain elusive. To understand this further, we review the literature on the τ and βA proteins and their breakdown, examine current treatments that look to only mask the symptoms, and introduce the idea that steroid hormones may provide a pathway to slowing down and possibly halting the progression of this disease. 2. Current Treatments Over the past couple of decades, many treatments have become the standard of care for AD patients. Medications range in the treatment for cognitive symptoms, behavioral control and insomnia [18] [19] [20] [21]. This is quite dramatic since there is not current medication or treatment that can reverse or stop AD from progressing. Therefore, treatments can only hope to slow down the appearance and progression of symptoms for a limited time. Regarding cognitive symptoms, cholinesterase inhibitors (Aricept, Exelon, Razadyne) and memantine (Namenda) have been used to treat the memory loss, confusion, and difficulties with thinking and reasoning of AD [22]. Cholinesterase inhibitors are typically used for mild to moderate case of AD, where they aim to reduce the breakdown of acetylcholine, which is an important chemical messenger that affects learning and memory. Therefore, these medications support communication among nerve cells by keeping acetylcholine levels high. The advantage of this kind of treatment is that it may delay worsening of symptoms for up to 12 months, which has been shown to affect about 50% the people who take them. However, the side effects of the medication have been severe, which include nausea, vomiting, loss of appetite and increased frequency of bowel movements [22]. To treat moderate to severe stages of AD, memantine may be prescribed. This medication affects the regulation of glutamate, which is similar to the cholinesterase inhibitors and works to delay symptoms of memory loss [23]. It is noted that the side effects tend to include a headache, constipation, confusion, and dizziness, at a minimum. T. S. Wiley, J. T. Haraldsen 35 Along with the cognitive symptoms, patients experience mood changes and other issues such as anxiety and depression [24]. This leads the patients to need a variety of antidepressant, anxiolytic, and antipsychotic medications. Overall, patients with AD are typically overwhelmed by so many medications and prescriptions that often interact. Beyond these pharmaceutical treatments, there are a number of alternative or complementary treatments that “claim” to have an effect on AD with no clinical evidence. These treatments include herbal remedies, dietary supplements, and food regimens. However, the effectiveness and safety of these treatments are also unknown, and they purveyors provide no known mechanisms of action for effectiveness. 3. The τ Hyperphosphorylation and β-Amyloid Plaques Tau proteins work to stabilize and maintain tubular polymers called microtubules that are abundant in the nerve cells of central nervous system [10]. τ proteins control microtubule stability through the use of its isoforms and phosphorylation [25]. Tau proteins are produced through a process of alternative splicing of a single gene called microtubule-associated protein τ [13]. Hyperphosphorylation of τ proteins can lead to the failure of microtubule stability, which can cause neurofibrillary tangles. When in combination with the formation of β-Amyloid plaques, this breakdown of biomechanisms are widely linked to Alzheimer’s disease [14]. Microtubules consist of long tubular structures that are assembled by protofilaments, which produced by tubulin α-β-dimers [26]. While these structures are dynamically stable, changes in the overall environment can affect the microtubules and cause them to breakdown. Since the τ protein controls this process, understanding how external and internal stressors influence the τ protein is relevant. In general, many stressors that could be affecting the τ protein include external (pollutants, electromagnetic fields, etc.) and internal (hormones, chemical intake, etc.) environmental factors [27]. Typically, it is thought that the increase in the τ hyperphosphorylation, which reduces the stability of the microtubules, causes β-amyloid plaques to form. However, recent research has suggested that it is the in βA proteins enhance hyperphosphorylation of the τ protein through glycogen synthase kinase 3 (GSK3) [28]. Therefore, as illustrated in Figure 1, the combination of the βA protein build up, and the breaking of the microtubule stability produce the creation of calcium plaques (βAP). This introduces inflammation around the neuron that can lead to transient ischemic attacks in the brain, which could cause permanent damage to memory and cognitive function. In recent years, AD and degeneration have been connected to the dysfunction of the mitochondria and the production of oxidative stress [7] [8] [15] [29], which is speculated to occur through formation of oxidative and nitrosative stresses, depletion of adenosine triphosphate, and impaired electron transport [7]. However, these subsequent dysfunctions are characteristic of aging effects due to loss T. S. Wiley, J. T. Haraldsen 36 Figure 1. General schematic of the formation of β-amyloid plaques due to decreased estrogen (E2) and τ protein. The right side illustrates the expected response to increased estrogen dosing. of hormones and its effect on mitochondria in both women and men, which has been widely documented in the literature [30]-[36]. This is why we focus on the effects produced by steroid hormones due to their ability to control and regulate gene protein products. 4. Effects of Steroid Hormones on Protein Expression The potential for the onset of AD is associated with the time in life when we experience the loss of sex steroid hormones in both women and men [17] [37] [38] [39]. In particular, studies on women show the decline of estrogen (E2) and absence of progesterone (P4) has been suggested to increase susceptibility to AD compared to men. Therefore, there are two possibilities: 1) the sharp decrease in neurological responsiveness in old age is due to the missing steroid hormones, or 2) the loss of steroid neurological feedback to the endocrine function destroys hormone production and brain responsiveness [40]. Experimental evidence has pointed to the promotion of neuron viability and the possible decrease in β-amyloid peptides due to the protective actions of E2 and the immunosuppressive action of P4 [41], which are a critical factor in the origination and advancement of AD and the neurological damage that can ensue. This connection suggests that the use of hormone replacement therapy (HRT) could provide a reduction in the risk of the onset of AD, but also perhaps decreases in AD symptoms. Furthermore, cyclic exposure to P4 helps in the regulation of hippocampal gene expression [42], which indicates the general effects of menopause on the reduction of gene expressions that can lead to diseases of aging. Over the last decade, there have been many studies that have suggested the re- T. S. Wiley, J. T. Haraldsen 37 levance of HRTs in the fight against and prevention of AD [31]-[36]. While these studies seem to demonstrate a significant reduction in risk of AD in women, other studies have suggested that the protective benefits of HRT may be overstated as some studies indicate little to no associated reduction in AD [43]-[48]. However, the dosing type in all of these studies was static synthetic estrogen and low in dosing amount. This means the women were given enough synthetic estrogen to elevate the overall concentration of hormones in the blood, but not enough to trigger the production of progesterone receptors [48]. Furthermore, they seem to completely ignore the standard bio-rhythmic response of the body that functions to control gene expression [49]. The regular biological rhythm for women has an increase in E2 during the first 12 days of a women’s cycle (illustrated in Figure 2). This peak in estrogen signals the production of P4 receptors, which, over a 9-day period, E2 is decreased in the body while P4 is increased [50]. Progesterone will peak on day 21, and then the cycle will reset on Day 28. It is this rhythmic pattern that is usually neglected in studies, with adverse outcomes, on HRTs for Alzheimer’s patients. We suspected that wide variation between effective and non-effective hormone treatments in the above studies is due to regularity of dosages, the delivery system (orally, transdermally, etc.), and/or whether progesterone is included at all in the treatment. As discussed in Pike et al. [30], estrogen’s neuro-protective actions are modulated by progesterone [51] [52], whereas static progesterone exposure always inhibits estrogen’s actions, synthetic or otherwise. However, the cyclic delivery of P4 and E2 will allow for the increase of E2 receptors [48] [49]. When low levels of E2 are administered, the potential production of progesterone receptors (P4R) is hindered [53] [54]. This can be improved through a rhythmic pattern of E2 and P4. Furthermore, estrogen is known to increase dendritic spining as well as synaptic plasticity [55] [56]. As observed in most women, the loss or disruption of E2 and P4 has a number Figure 2. The suggested rhythmic dosing pattern for estrogen (E2) and progesterone (P4) during the normal reproductive cycle for a young woman. As E2 peaks, so does the production of P4 receptors. Around Day 14, women will experience a peak in luteinizing hormone (LH) and follicle-stimulating hormone (FSH), which begin the ovulation process. T. S. Wiley, J. T. Haraldsen 38 of effects on the body and mind. For example, decreased E2 will reduce synaptic plasticity and increase the loss of dendritic spinning [55] [56]. The disruption of the production of progesterone and P4 receptors, via luteinizing hormone (LH), due to estrogen loss produces diminished immunosuppression and vascular endothelial growth factor. Furthermore, it is well documented that the presence of estrogen prevents hyperphosphorylation of the τ protein and helps regulate the metabolism of β-amyloid. Therefore, it is probable that reduction or complete loss of estrogen and progesterone can lead to increased hyperphosphorylation of τ (illustrated in Figure 3) and an increase in β-amyloid, which, in turn, also reinforces hyperphosphorylation resulting in the onset of AD. Since the loss of E2 concurrently produces a loss of P4, the body will experience diminished immunosuppression and loss of Vascular Endothelial Growth Factor (VEGF) [57]. Therefore, we propose that the effect of rhythmic dosing of estrogen with dosing levels comparable to those of a reproductive woman may reduce hyperphosphorylation of τ and subsequently inflammation around calcium plaques (illustrated in Figure 1), which may decrease the amount of memory loss produced from multiple random transient ischemic attacks (as shown in the right side of Figure 1). The effects of E2 restoration should increase the brain’s ability to increase dendritic spining and synaptic plasticity, which may lead to a decrease in the formation of plaques; although this would have to be studied clinically. Furthermore, the presence of E2 above the critical threshold produces P4 receptors. The levels of rhythmic P4 will restore that steroid hormone to youthful levels, which will create an increase in immunosuppressive response and VEGF, which has been shown to help restore memory behavior in mice [58], Figure 3. Generalization chain of events for loss of steriods hormones (estrogen-E2 and progesterone-P4) and the effect it has on Alzheimer’s disease. T. S. Wiley, J. T. Haraldsen 39 and helps with the reduction of inflammation around calcium plaques. The increasing dendritic spining and loss of τ hyperphosphorylation may also help remove plagues, but this is purely speculative. 5. Conclusions Through an evaluation of current literature, we examine the possible effects of the steroid hormones (specifically E2 and P4) on the regulation of τ hyperphosphorylation and β-amyloid and their influence on the formation of β-amyloid plaques in Alzheimer’s patients. We present a general mechanism that Alzheimer’s cases may be reduced or even prevented with the use of non-synthetic steroid hormones prescribed in a cyclic dosing schedule which mimics the rhythmic, escalating, and descending production in a reproductive body to control gene expression. Given the ability of E2 to prevent τ hyperphosphorylation and to regulate βA, there is a possible method for controlling hyperphosphorylation and β-amyloid plaque formation through the application of estrogen and progesterone may help those patients with active disease as well as prevent the onset of Alzheimer’s, if begun before menopause. Furthermore, using a bio-mimetic hormone replacement methodology, we examine and propose that modulated hormone dosing and the effects of steroid pulsatility and amplitude at the receptor level are logically a major factor in effectively controlling the expression in the brain. Future aspects of research include examining how bio-mimetic, bio-identical hormone replacement therapy affects the maintenance of microtubules and the production of τ and β-amyloid. We propose a clinical experiment that will administer bio-identical hormones to women in a manner which mimics the normal rhythmic patterns of young reproductive women. The hormones should be administered using a transdermal cream. This allows for the hormones to be more effective, since they will bypass the liver. As a control, this should be compared to other hormone therapies (variable and static timing), as well as the control group with no hormones (transdermal cream without hormones). Investigations into the advancement of AD can be determined through standard medical techniques (magnetic resonance imaging and CT scanning) along with physical, diagnostic, and neurological examinations. As per the mechanism above, we expect that there will be a slowing or abatement of AD symptoms due to the restoration of youthful gene products. A reversal of symptoms may be theoretically possible, but other factors such as damage may hinder those possibilities. Once a satisfactory baseline can be established for women, further investigation into the estrogen derived from testosterone in men may also be studied. While the mechanism for men is not clear, due to the lack of a distinct hormonal pattern, the increase of testosterone will increase the presence of estrogen in the body and may have the same effect overall. However, this exact mechanism is not evident at this time. Overall, as people age, the only one constant among all people is that we lose T. S. Wiley, J. T. Haraldsen 40 the ability to produce sex steroid hormones and regulate the proper production and regulation of gene products at their behest. It is our conclusion that the manifestation of Alzheimer’s disease may be due to this particular problem. In women, the loss of E2 and P4 produces a cascade of events that can result in the growth of calcium plaques on neurons and the subsequent production of transient ischemic attacks that lead to memory loss. The same may also be true for men, although through the loss of testosterone, which typically converts to estrogen. However, when looking at a problem with so many complex interacting parts, it is sometimes needed to step back and acknowledge the loss of something that is basic to human growth and reproduction as the possible cause. Acknowledgements T. S. W. and J. T. H. acknowledge useful and insightful discussions with Mara Raden and Victor Rosenfeld. References [1] Ouslander, J.G., Studenski, S. and Halter, J.B. (2009) Hazzard’s Geriatric Medicine and Gerontology. 6th Edition, The McGraw-Hill Companies, New York. [2] Katzman, R. and Saitoh, T. (1991) Advances in Alzheimer’s Disease. The FASEB Journal, 5, 278. [3] Mann, D.M. and Esiri, M.M. (1989) The Pattern of Acquisition of Plaques and Tangles in the Brains of Patients under 50 Years of Age with Down’s Syndrome. Journal of the Neurological Sciences, 89, 169-179. https://doi.org/10.1016/0022-510X(89)90019-1 [4] Selkoe, D.J. (1991) The Molecular Pathology of Alzheimer’s Disease. Neuron, 6, 487. https://doi.org/10.1016/0896-6273(91)90052-2 [5] Blessed, G., Tomlinson, B.E. and Roth, M. (1968) The Association between Quantitative Measures of Dementia and of Senile Change in the Cerebral Grey Matter of Elderly Subjects. The British Journal of Psychiatry, 114, 797. https://doi.org/10.1192/bjp.114.512.797 [6] Armstrong, R. (2013) What Causes Alzheimer’s Disease? Folia Neuropathologica, 5, 169-188. https://doi.org/10.5114/fn.2013.37702 [7] Bhat, A.H., Dar, K.B., Anees, S., Zargar, M.A., Masood, A., Sofi, M.A., et al. (2015) Review: Oxidative Stress, Mitochondrial Dysfunction and Neurodegenerative Diseases: A Mechanistic Insight. Biomedicine & Pharmacotherapy, 74, 101-110. https://doi.org/10.1016/j.biopha.2015.07.025 [8] Castellani, R., Hirai, K., Aliev, G., Drew, K.L., Nunomura, A., Takeda, A., et al. (2002) Role of Mitochondrial Dysfunction in Alzheimer’s Disease. Journal of Neuroscience Research, 70, 357-360. https://doi.org/10.1002/jnr.10389 [9] Maccioni, R.B., Muñoz, J.P. and Barbeito, L. (2001) The Molecular Bases of Alzheimer’s Disease and other Neurodegenerative Disorders. Archives of Medical Research, 32, 367-381. https://doi.org/10.1016/S0188-4409(01)00316-2 [10] Shin, R.W., Iwaki, T., Kitamoto, T. and Tateishi, J. (1991) Hydrated Autoclave Pretreatment Enhances Tau Immunoreactivity in Formalin-Fixed Normal and Alzheimer’s Disease Brain Tissues. Laboratory Investigation, 64, 693-702. [11] Lei, P., Ayton, S., Finkelstein, D.I., Adlard, P.A., Masters, C.L. and Bush, A.I. (2010). Tau Protein: Relevance to Parkinson’s Disease. The International Journal of Bioche- T. S. Wiley, J. T. Haraldsen 41 mistry & Cell Biology, 42, 1775-1778. https://doi.org/10.1016/j.biocel.2010.07.016 [12] Brier, M.R., Gordon, B., Friedrichsen, K., McCarthy, J., Stern, A., Christensen, J., Owen, C., Aldea, P., Su, Y., Hassenstab, J., Cairns, N.J., Holtzman, D.M., Fagan, A.M., Morris, J.C., Benzinger, T.L.S. and Ances, B.M. (2016) Tau and Ab Imaging, CSF Measures, and Cognition in Alzheimer’s Disease. Science Translational Medicine, 8, 338. https://doi.org/10.1126/scitranslmed.aaf2362 [13] Weingarten, M.D., Lockwood, A.H., Hwo, S.Y. and Kirschner, M.W. (1975) A Protein Factor Essential for Microtubule Assembly. PNAS, 72, 1858-1862. https://doi.org/10.1073/pnas.72.5.1858 [14] Goedert, M., Wischik, C.M., Crowther, R.A., Walker, J.E. and Klug, A. (1988) Cloning and Sequencing of the cDNA Encoding a Core Protein of the Paired Helical Filament of Alzheimer Disease: Identification as the Microtubule-Associated Protein Tau. PNAS, 85, 4051-4055. https://doi.org/10.1073/pnas.85.11.4051 [15] Reddy, P.H. and Beal, M.F. (2008) Amyloid Beta, Mitochondrial Dysfunction and Synaptic Damage: Implications for Cognitive Decline in Aging and Alzheimer’s Disease. Trends in Molecular Medicine, 14, 45-53. https://doi.org/10.1016/j.molmed.2007.12.002 [16] Henderson, V.W., Paganini-Hill, A., Emanuel, C.K., Dunn, M.E. and Buckwalter, J.G. (1994) Estrogen Replacement Therapy in Older Women. Comparisons between Alzheimer’s Disease Cases and Nondemented Control Subjects. Archives of Neurology, 51, 896-900. https://doi.org/10.1001/archneur.1994.00540210068014 [17] Henderson, V.W. (1997) Estrogen, Cognition, and a Woman’s Risk of Alzheimer’s Disease. The American Journal of Medicine, 103, 11S-18S. https://doi.org/10.1016/S0002-9343(97)00261-1 [18] Kelly, C.A., Harvey, R.J. and Cayton, H. (1997) Drug Treatments for Alzheimer’s Disease. British Medical Journal, 314, 693-694. https://doi.org/10.1136/bmj.314.7082.693 [19] Corbett, A., Smith, J. and Ballard, C. (2012) New and Emerging Treatments for Alzheimer’s Disease. Expert Review of Neurotherapeutics, 12, 535-543. https://doi.org/10.1586/ern.12.43 [20] Wilkerson, D. (2001) Drugs for Treatment of Alzheimer’s Disease. International Journal of Clinical Practice, 55, 129-134. [21] Max, W. (1999) Drug Treatments for Alzheimer’s Disease. CNS Drugs, 11, 363-372. https://doi.org/10.2165/00023210-199911050-00004 [22] Doody, R.S. (2003) Current Treatments for Alzheimer’s Disease: Cholinesterase Inhibitors. The Journal of Clinical Psychiatry, 64, 11-17. [23] Reisberg, B., Doody, R., Stöffler, A., Schmitt, F., Ferris, S. and Möbius, H.J. (2003) Memantine in Moderate-to-Severe Alzheimer’s Disease. New England Journal of Medicine, 348, 1333-1341. https://doi.org/10.1056/NEJMoa013128 [24] Thompson, S., Herrmann, N., Rapoport, M.J. and Lanctôt, K.L. (2007) Efficacy and Safety of Antidepressants for Treatment of Depression in Alzheimer’s Disease: A Meta-Analysis. The Canadian Journal of Psychiatry, 52, 248-255. https://doi.org/10.1177/070674370705200407 [25] Harada, A., Oguchi, K., Okabe, S., Kuno, J., Terada, S., Ohshima, T., et al. (1994) Altered Microtubule Organization in Small-Calibre Axons of Mice Lacking Tau Protein. Nature, 369, 488-491. https://doi.org/10.1038/369488a0 [26] Howard, J. and Hyman, A.A. (2007) Microtubule Polymerases and Depolymerases. Current Opinion in Cell Biology, 19, 31-35. T. S. Wiley, J. T. Haraldsen 42 https://doi.org/10.1016/j.ceb.2006.12.009 [27] Stoothoff, W.H. and Johnson, G.V.W. (2005) Tau Phosphorylation: Physiological and Pathological Consequences. Biochimicaet Biophysica Acta―Molecular Basis of Disease, 1739, 280-297. https://doi.org/10.1016/j.bbadis.2004.06.017 [28] Hernández, F., Gómezde Barreda, E., Fuster-Matanzo, A., Lucas, J.J. and Avila, J. (2010) GSK3: A Possible Link between Beta Amyloid Peptide and Tau Protein. Experimental Neurology, 223, 322-325. https://doi.org/10.1016/j.expneurol.2009.09.011 [29] Lin, M.T. and Beal, M.F. (2006) Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature, 443, 87-95. https://doi.org/10.1038/nature05292 [30] Pike, C.J., Carroll, J.C., Rosario, E.R. and Barron, A.M. (2009) Protective Actions of Sex Steroid Hormones in Alzheimer’s Disease. Front Neuroendocrinol, 30, 239-258. https://doi.org/10.1016/j.yfrne.2009.04.015 [31] Kawas, C., Resnick, S., Morrison, A., Brookmeyer, R., Corrada, M., Zonderman, A., et al. (1997) A Prospective Study of Estrogen Replacement Therapy and the Risk of Developing Alzheimer’s Disease: The Baltimore Longitudinal Study of Aging. Neurology, 48, 1517-1521. https://doi.org/10.1097/00006254-199711000-00020 [32] Paganini-Hill, A. and Henderson, V.W. (1996) Estrogen Replacement Therapy and Risk of Alzheimer’s Disease. Archives of Internal Medicine, 156, 2213-2217. https://doi.org/10.1001/archinte.1996.00440180075009 [33] Tang, M.X., Jacobs, D., Stern, Y., Marder, K., Schofield, P., Gurland, B., et al. (1996) Effect of Oestrogen during Menopause on Risk and Age at Onset of Alzheimer’s Disease. The Lancet, 348, 429-432. https://doi.org/10.1016/S0140-6736(96)03356-9 [34] Waring, S.C., Rocca, W.A., Petersen, R.C., O’Brien, P.C., Tangalos, E.G. and Kokmen, E. (1999) Postmenopausal Estrogen Replacement Therapy and Risk of AD: A Population-Based Study. Neurology, 52, 965-970. https://doi.org/10.1212/WNL.52.5.965 [35] Zandi, P.P., Carlson, M.C., Plassman, B.L., Welsh-Bohmer, K.A., Mayer, L.S., Steffens, D.C., et al. (2002) Hormone Replacement Therapy and Incidence of Alzheimer Disease in Older Women: The Cache County Study. JAMA, 288, 2123-2129. https://doi.org/10.1001/jama.288.17.2123 [36] Carlson, M.C., Zandi, P.P., Plassman, B.L., Tschanz, J.T., Welsh-Bohmer, K.A., Steffens, D.C., et al. (2001) Hormone Replacement Therapy and Reduced Cognitive Decline in Older Women: The Cache County Study. Neurology, 57, 2210-2216. https://doi.org/10.1212/WNL.57.12.2210 [37] Rosario, E.R., Chang, L., Head, E.H., Stanczyk, F.Z. and Pike, C.J. (2011) Brain Levels of Sex Steroid Hormones in Men and Women during Normal Aging and in Alzheimer’s Disease. Neurobiology of Aging, 32, 604-613. https://doi.org/10.1016/j.neurobiolaging.2009.04.008 [38] Hu, X.-Y., Qin, S., Lu, Y.-P., Ravid, R., Swaab, D.F. and Zhou, J.-N. (2003) Decreased Estrogen Receptor-Alpha Expression in Hippocampal Neurons in Relation to Hyperphosphorylated Tau in Alzheimer Patients. Acta Neuropathologica, 106, 213-220. https://doi.org/10.1007/s00401-003-0720-3 [39] Alvarez-De-La-Rosa, M., Silva, I., Nilsen, J., Pérez, M.M., García-Segura, L.M., Ávila, J., et al. (2005) Estradiol Prevents Neural Tau Hyperphosphorylation Characteristic of Alzheimer’s Disease. Annals of the New York Academy of Sciences, 1052, 210-224. https://doi.org/10.1196/annals.1347.016 [40] Galea, L.A.M., Frick, K.M., Hampson, E., Sohrabji, F. and Choleris, E. (2016) Why Estrogens Matter for Behavior and Brain Health. Neuroscience & Biobehavioral Re- T. S. Wiley, J. T. Haraldsen 43 views. (In Press) https://doi.org/10.1016/j.neubiorev.2016.03.024 [41] Xu, Y., He, H., Li, C., Shi, Y., Wang, Q., Li, W., et al. (2011) Immunosuppressive Effect of Erogesterone on Dendritic Cells in Mice. Journal of Reproductive Immunology, 91, 17-23. [42] Zhao, L., Morgan, T.E., Mao, Z., Lin, S., Cadenas, E., Finch, C.E., et al. (2012) Continuous versus Cyclic Progesterone Exposure Differentially Regulates Hippocampal Gene Expression and Functional Profiles. PLoS ONE, 29, 7. [43] Haskell, S.G., Richardson, E.D. and Horwitz, R.I. (1997) The Effect of Estrogen Replacement Therapy on Cognitive Function in Women: A Critical Review of the Literature. Journal of Clinical Epidemiology, 50, 1249-1264. https://doi.org/10.1016/S0895-4356(97)00169-8 [44] Almeida, O.P., Lautenschlager, N.T., Vasikaran, S., Leedman, P., Gelavis, A. and Flicker, L. (2006) A 20-Week Randomized Controlled Trial of Estradiol Replacement Therapy for Women Aged 70 Years and Older: Effect on Mood, Cognition and Quality of Life. Neurobiology of Aging, 27, 141-149. https://doi.org/10.1016/j.neurobiolaging.2004.12.012 [45] Barrett-Connor, E. and Kritz-Silverstein, D. (1993) Estrogen Replacement Therapy and Cognitive Function in Older Women. JAMA, 269, 2637-2641. https://doi.org/10.1001/jama.1993.03500200051032 [46] Brenner, D.E., Kukull, W.A., Stergachis, A., van Belle, G., Bowen, J.D., McCormick, W.C., et al. (1994) Postmenopausal Estrogen Replacement Therapy and the Risk of Alzheimer’s Disease: A Population-Based Case-Control Study. American Journal of Epidemiology, 140, 262-267. https://doi.org/10.1093/oxfordjournals.aje.a117245 [47] Goebel, J.A., Birge, S.J., Price, S.C., Hanson, J.M. and Fishel, D.G. (1995) Estrogen Replacement Therapy and Postural Stability in the Elderly. American Journal of Otolaryngology, 16, 470-474. [48] Wiley, T.S. and Haraldsen, J.T. (2012) The Theory of Modulated Hormone Therapy for the Treatment of Breast Cancer in Pre- and Post-Menopausal Women. AIP Advances, 2, Article ID: 011206. https://doi.org/10.1063/1.3699052 [49] Wiley, T.S. and Haraldsen, J.T. (2014) The Effects of Steroid Hormone Exposure on Direct Gene Regulation. Medical Hypotheses, 83, 436-440. https://doi.org/10.1016/j.mehy.2014.07.010 [50] Vande Wiele, R.L., Bogumil, J., Dyrenfurth, I., Ferin, M., Jewelewicz, R., Warren, M., et al. (1970) Mechanisms Regulating the Menstrual Cycle in Women. Recent Progress in Hormone Research, 126, 63-103. [51] Pozzi, S., Benedusi, V., Maggi, A. and Vegeto, E. (2006) Estrogen Action in Neuroprotection and Brain Inflammation. Annals of the New York Academy of Sciences, 1089, 302-323. https://doi.org/10.1196/annals.1386.035 [52] Vegeto, E., Benedusi, V. and Maggi, A. (2008) Estrogen Anti-Inflammatory Activity in Brain: A Therapeutic Opportunity for Menopause and Neurodegenerative Diseases. Frontiers in Neuroendocrinology, 29, 507-519. https://doi.org/10.1016/j.yfrne.2008.04.001 [53] Horwitz, K.B. and McGuire, W.L. (1978) Estrogen Control of Progesterone Receptor in Human Breast Cancer. The Journal of Biological Chemistry, 253, 2223-2228. [54] Chen, T.J. and Jow, G.M. (1989) Effects of Estrogen on Progesterone Receptor Synthesis during Endometrial Cell Culture. Journal of the Formosan Medical Association, 88, 456-461. [55] Segal, M. and Murphy, D. (2001) Estradiol Induces Formation of Dendritic Spines in Hippocampal Neurons: Functional Correlates. Hormones and Behavior, 40, 156- T. S. Wiley, J. T. Haraldsen 44 159. https://doi.org/10.1006/hbeh.2001.1688 [56] Stuart, G., Spruston, N. and Häusser, M. (2016) Dendrites. Oxford University Press, Oxford, 642 p. https://doi.org/10.1093/acprof:oso/9780198745273.001.0001 [57] Kalaria, R.N., Cohen, D.L., Premkumar, D.R., Nag, S., LaManna, J.C. and Lust, W.D. (1998) Vascular Endothelial Growth Factor in Alzheimer’s Disease and Experimental Cerebral Ischemia. Brain Research. Molecular Brain Research, 62, 101-105. https://doi.org/10.1016/S0169-328X(98)00190-9 [58] Religa, P., Cao, R., Religa, D., Xue, Y., Bogdanovic, N., Westaway, D., et al. (2013) VEGF Significantly Restores Impaired Memory Behavior in Alzheimer’s Mice by Improvement of Vascular Survival. Scientific Reports, 3, 2053.

 发给朋友
发给朋友 分享到朋友圈
分享到朋友圈 回顶部
回顶部